摘要
乳腺癌已成为全球女性发病率最高且发病率呈上升趋势的恶性肿瘤。化疗、内分泌治疗及分子靶向治疗等传统治疗手段在临床上取得了一定成效,但其耐药性问题严重影响了治疗效果。蛋白降解靶向嵌合体(Proteolysis Targeting Chimeric,PROTAC)技术作为一种新兴的蛋白质降解策略,为治疗耐药性乳腺癌提供了新的可能性,其中天然产物衍生的PROTAC分子展现出巨大潜力。该文总结了PROTAC技术在乳腺癌治疗中的应用现状、不同分子分型乳腺癌的关键靶点PROTAC的研究进展,探讨基于天然产物的PROTAC分子设计及其在乳腺癌治疗中的潜在价值及基于 PROTAC技术开发的海洋药物在乳腺癌治疗中的研究前景。
Abstract
Breast cancer has become the most prevalent malignancy among women globally, with an increasing incidence rate. Traditional treatment modalities such as chemotherapy, endocrine therapy, and molecular targeted therapy have achieved certain clinical successes, but their effectiveness is severely compromised by drug resistance issues. Proteolysis Targeting Chimeric (PROTAC) technology, as an emerging protein degradation strategy, offers new possibilities for treating drug-resistant breast cancer, particularly PROTAC molecules derived from natural products exhibited great potential. This review summarized the current status and future trends of PROTAC technology in breast cancer treatment. It focuses on PROTAC research progress targeting different molecular subtypes of breast cancer, delves into the design of PROTAC molecules based on natural products, and explores their potential value in breast cancer treatment. Additionally, the review provides new insights into the prospects of marine drugs developed based on PROTAC technology in breast cancer treatment.
Keywords
乳腺癌是全球女性最常患的恶性肿瘤,发病率不断攀升,严重危及女性生命健康。传统化疗、内分泌治疗与分子靶向治疗虽有成效,但耐药性问题限制了疗效提升[1]。本文系统梳理蛋白降解靶向嵌合体(PROTAC)技术及其天然产物衍生分子在乳腺癌治疗方面的现状与发展走向,以期对基于PROTAC技术开发海洋药物在乳腺癌治疗中的设计提供参考。
1 乳腺癌治疗现状
在乳腺癌分子病理学诊断中,根据雌激素受体 (estrogen receptor,ER)、孕激素受体(progesterone receptor,PR)、人表皮生长因子受体-2(human epidermal growth factor receptor-2,HER-2)表达水平不同,乳腺癌可粗略分为 3 类:激素受体阳型(ER+)、HER-2 阳型(HER-2+)和三阴型乳腺癌(TNBC)[1]。大约 70% 的乳腺癌严重依赖ER触发因子来促进肿瘤生长和存活[2]。用于治疗ER+的乳腺癌药物,包括芳香酶抑制剂(AI)、选择性ER调节剂(SERM)和选择性ER降解剂(SERD)等均有明显疗效,但其原发性和继发性耐药仍然是威胁乳腺癌群体生命安全的隐患。HER-2+ 乳腺癌通过大分子单克隆抗体、小分子酪氨酸激酶抑制剂或抗体偶联药物阻止人体表皮生长因子在HER-2 上的附着,从而阻断癌细胞的生长。相较于 ER+ 和 HER-2+亚型,TNBC缺乏特异性表达,是目前乳腺癌治疗中最棘手的挑战之一,其常规治疗仍是细胞毒药物,缺乏FDA批准的针对TNBC的靶向药物[3]。
2 PROTAC技术研究现状
2001 年,Craig Crews 教授及其团队首次提出了 PROTAC 的概念 [4]。PROTAC 是包含两个不同配体的异双功能分子,其中一头用于结合目标蛋白(POI),另一头用于结合E3 连接酶。其原理是将E3 连接酶引入POI区域,通过E3 连接酶进行泛素化,并随后进行蛋白酶体分解[5]。图1显示了PROTAC降解过程: PROTAC穿透细胞,与E3 连接酶和POI作用,实现POI 降解。进膜后,PROTAC结合POI和E3 连接酶,催化多聚泛素化,由蛋白酶体降解。E3 将泛素从E2 复合物移至POI,重复形成多聚泛素化POI,被蛋白酶体识别降解。三元络合物稳定时,Ub分子移动至POI并泛素化,降解POI以达疗效。即PROTAC由 1 个与E3 泛素连接酶结合的配体和 1 个通过接头与靶蛋白结合的配体组成,可诱导细胞内靶蛋白的多聚泛素化和蛋白酶体降解。PROTAC展现了一种相较于传统方法而言,独树一帜的活性机制[6]。其核心优势在于,PROTAC 介导的蛋白质降解过程是 1 个高度事件驱动的过程,这与传统药物通过占位驱动来影响蛋白质功能的模式截然不同。传统药物通常需长时间占据特定的生物活性位点以发挥药效,而PROTAC只需提供结合活性,触发靶蛋白与E3 酶结合从而引发降解。这种特性使得它能够实现更为高效的靶标降解,有效规避耐药性的问题。理论上讲,仅需催化量的PROTAC即可预期引发靶蛋白显著减少,这一需求量相较于传统药物而言更为科学合理[7]。因此,在面对恶性肿瘤的耐药性增长问题时,PROTAC展现出了非凡的潜力[8]。
PROTAC 领域的研究焦点主要集中在癌症治疗应用上。现阶段,已有数十种PROTAC药物正处于临床试验阶段。其中,于 2015 年第一个进入临床试验的 PROTAC 药物 ARV-110 现已迈入治疗前列腺癌 Ⅱ 期临床试验进程,而同样针对前列腺癌的ARV-766、CC-94676、AC-0176 以及 HP518 等药物则正处于 Ⅰ 期临床试验阶段。值得注意的是,2024 年 2 月,利用天然内源性激素 17β-雌二醇(E2)作为ER的弹头制备而成的ARV-471 已被FDA授予单药治疗快速通道资格,用于治疗既往接受过内分泌治疗的ER+、HER-2 局部晚期或转移性乳腺癌患者。至此,PROTAC技术为解决耐药问题提供了一种很有前景的策略[9]。

图1PROTAC介导的POI泛素化和蛋白酶体降解过程
3 天然产物研究现状
天然产物是抗癌药物和先导化合物的主要来源[10]。与化学合成的分子相比,天然产物凭借其独特的结构特征、优越的生物相容性和多功能性[11-12],在药物发现领域持续展现出不可替代的价值。然而,大多数天然产物与蛋白质靶点的相互作用往往仅表现出中等亲和力,这为其新药研发带来了复杂性与困难[13-14]。 PROTAC技术通过利用蛋白质表面嵌入式识别机制,使得天然产物及其衍生物即便在缺乏高亲和力的情况下,也能有效降解目标蛋白,从而在新药研发领域展现出巨大潜力。众多天然产物已被证实可通过特异性结合相关蛋白质靶点来发挥生物活性[15-16],因此具备作为 E3 连接酶或POI 连接酶的潜力。PROTAC 技术在天然产物领域的应用,将是对现有方法(如直接使用生物活性天然产物或其类似物、药效团开发以及药物-抗体偶联物)的有效补充。
4 针对乳腺癌的PROTAC分子
在乳腺癌的治疗领域,PROTAC技术正致力于探索通过直接去除致病靶蛋白来达成治疗目的、有效规避原发性和继发性耐药的新途径 [17]。随着研究的深入,越来越多的PROTAC分子被针对性地开发出来,用于治疗不同类型的乳腺癌。这些尝试不仅拓宽了乳腺癌的治疗手段,也为患者提供了新的希望。在具体作用机制上,PROTAC降解蛋白质涉及泛素连接酶酶促反应,使目标底物泛素化,泛素化目标底物可进一步被蛋白酶体识别,进而完成蛋白降解(图2),利用这一机制有利于开展对恶性肿瘤的治疗。以下汇总了一些针对乳腺癌治疗的PROTAC分子实例,展示了这一领域的研究进展与潜力。
4.1 靶向ER的PROTAC分子
与正常组织相比,ER(包括ERα和ERβ两种异构体)蛋白水平在癌前病变和恶性病变中差异较大,大约 70% 的乳腺癌严重依赖于 ER 来促进癌细胞的增殖和转移 [18]。他莫昔芬和氟维司群作为抗雌激素治疗药物,已广泛用于不同阶段ER+乳腺癌治疗,能够竞争性结合并抑制ER从而抑制细胞增殖[19-20]。然而,大多数初治疗效果良好的乳腺癌最终会发展出耐药性[21-22]。开发既能抑制激素应答性乳腺癌生长又能预防耐药性的新型治疗药物成为一大挑战[23]。

图2PROTAC在乳腺癌治疗中降解蛋白质的作用机制
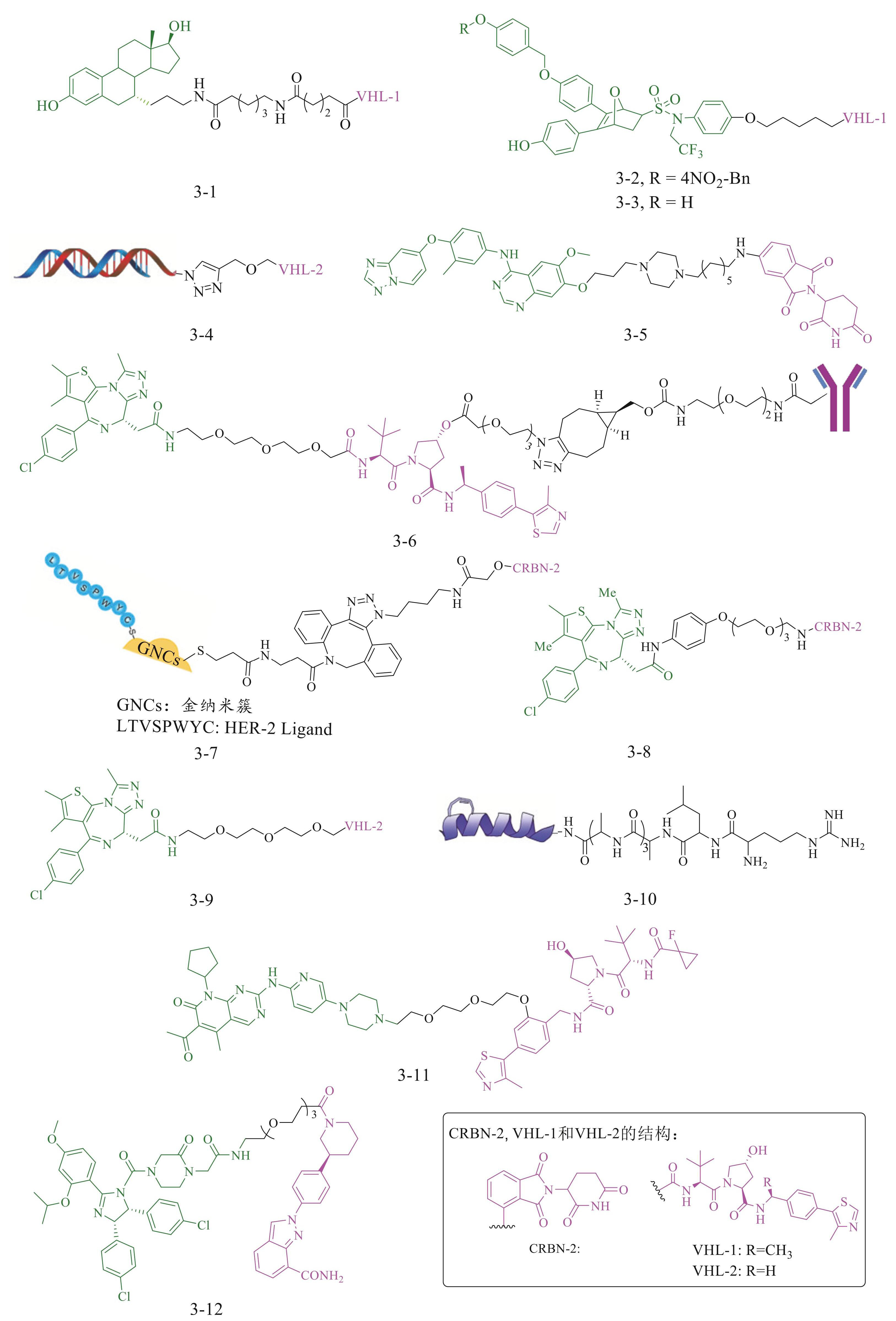
Cyrus 等 [24] 设计了一种靶向 ER 的 PROTAC,其结构的一端嵌入雌二醇,另一端则连接着源自HIF-1α 的合成五肽。通过将五肽整合至雌二醇分子的 3 个不同位点,他们成功合成了 3 种不同的PROTACs。其中,C7α-五肽 PROTAC(图3-1)展现出了卓越的ER 降解能力和受体亲和力。这些发现为开发能够克服乳腺癌对传统药物(例如他莫昔芬和氟维司群)耐药性的新型ER拮抗剂提供了可能。此外,鉴于肿瘤微环境的复杂性,Xie等[25]设计了一种在ER配体或E3 配体中融入低氧活化基团(如硝基咪唑和硝基苯)的ERα PROTAC(图3-2),从而提高了PROTAC在低氧条件下的活性和乳腺癌治疗的安全性。Xie等[25]还报道一组基于OBHSA支架构建的无ERβ降解活性的强效和高选择性ERα PROTAC。在对他莫昔芬敏感和耐药的小鼠乳腺癌模型中,化合物ZD12 PROTAC(图3-3) 展现出了优于氟维司群的抗肿瘤效果和 ERα 降解能力。为了规避由配体结合域(LBD)诱导的耐药性, Zhang等[7]选择了ERα的DNA结合域(DBD)作为新的靶点,利用天然的ERE序列与ERα的DBD相互作用,作为PROTAC的识别单元,并通过点击反应合成了核酸偶联的ERE PROTAC(图3-4)。Disch等[26]还构建了用于ERα WT和 3 种功能获得性突变体筛选的 DNA编码化学文库,发现了新的ERα PROTAC。这项研究从DNA编码化学库中筛选出新的双特异性降解 PROTAC,提供了简易设计PROTAC的新思路。
4.2 靶向HER-2 的PROTAC分子
在乳腺癌中,HER-2 不仅是重要的驱动基因和预后指标,也是抗HER-2 药物治疗效果的主要预测因子[27]。尽管抗HER-2 治疗在降低HER-2 阳性乳腺癌转移与复发风险方面取得了显著进展,但耐药性问题仍普遍存在[28]。Hu等[29]报道的HER-2 选择性CH7C4 PROTAC(图3-5)能够有效降解HER-2,作为首个体外和体内活性均优于图卡替尼(HER-2 抑制剂)的选择性HER-2 PROTAC降解剂,为开发HER-2 阳性乳腺癌的治疗提供了新策略。Maneiro等[30]则开发了一种曲妥珠单抗-PROTAC偶联物Ab PROTAC(图3-6),选择性靶向降解HER-2 阳性乳腺癌细胞系。该研究证明了组织特异性降解的概念,克服了PROTAC选择性的局限性,具有应用于新靶标的巨大潜力。基于肽的 PROTACs存在的细胞渗透性差、稳定性和效力低等缺点,Wang等[31]设计了一种纳米级GNCTACs PROTAC (图3-7),可靶向并降解肿瘤细胞中的HER-2。该设计利用金纳米团簇(GNCs)连接HER-2 靶向肽和小脑蛋白(CRBN)靶向配体,能够克服肽基PROTACs的内在障碍,有效在细胞质中传递HER-2 靶向肽并使其免受降解。
4.3 治疗TNBC的PROTAC分子
TNBC具有高度侵袭性,临床预后差,无靶向治疗,亟需探索新的治疗策略。由于TNBC缺乏ER和 HER-2 的表达,因此治疗 TNBC 的 PROTAC 被设计为靶向降解在TNBC细胞系中过表达的蛋白质,例如溴结构域和末端外蛋白(BET)、类固醇受体共激活因子-1(SRC-1)、细胞周期蛋白依赖性激酶(CDK)和多聚ADP核糖聚合酶 1(PARP 1)等。BET蛋白家族(包括 BRD2、BRD3、BRD4 和 BRDT)通过 BD1 和 BD2 特异性地结合组蛋白H3、H4 或非组蛋白中的N-ε-乙酰赖氨酸(KAc)残基,进而调控转录和染色质重塑。 Noblejas-López等[32]基于BET蛋白家族合成并评估了 ARV-825 PROTAC(图3-8)和MZ1 PROTAC(图3-9) 两种针对 BET 的 PROTACs 在 TNBC 中的抗肿瘤活性,发现这些PROTACs有效维持了MDA-MB-231 和耐药JQ1 细胞系中BRD4 的缺失水平。在TNBC中,高水平的 SRC-1 表达与患者预后不良有关 [33]。Lee 等[34]利用UBR E3 连接酶和SRC-1 的结合伴侣STAT6 开发了一种 SRC-1 降解剂 ND1-YL2 PROTAC(图3-10),发现 ND1-YL 2 PROTAC 在体外和体内抑制 MDA-MB-231 TNBC侵袭和迁移。异常的CDK活化导致细胞增殖失调,这是癌症的一个关键病理机制[35]。为避免基于CRBN的CDK 4/6 PROTAC存在的CRBN 失活等因素导致的耐药性,Steinebach等[36]制备了对 CDK 4 和 CDK 6 都表现出有效的降解能力的 CDK 4/6 PROTAC(图3-11),同时在不改变VHL、IKZF1 或 IKZF3 的水平的情况下,对乳腺癌细胞系具有显著的抑制作用。Li等[37]基于PARP1 进一步开发了一种基于合成致死性机制的NN3 PROTAC(图3-12),用于降解TNBC中因PARP1 蛋白点突变而导致的PARPis获得性耐药。
4.4 基于天然产物治疗乳腺癌的PROTAC分子
自 2001 年开发了基于天然鹅膏菌素 1 的PROTAC(图4-1) 用于降解甲硫氨酸氨肽酶-2 (MetAP-2)以来,PROTAC技术在天然产物研究与开发领域的应用便备受瞩目 [4]。2024 年,FDA 批准的 ARV-471(图4-2)作为治疗接受过内分泌治疗的ER 阳性、HER-2 阴性局部晚期或转移性乳腺癌患者的 PROTAC降解剂,标志着这一技术的重大突破。该药物利用天然内源性激素 17β-雌二醇(E2)作为ER的弹头制备而成[38]。土槿皮酸B(PAB),一种源自日本土槿皮根和树干的三环二萜类化合物[39],对包括乳腺癌在内的多种肿瘤生长具有显著抑制作用[40-42]。Zhou 等[43]设计并合成了首批基于PAB的PROTAC(图4-3),通过连接PAB和CRBN配体实现了对CD147(PAB的靶蛋白)的靶向降解,为致癌CD147 蛋白的降解提供了一种可行方法,并展示了基于PAB的PROTAC作为癌症治疗药物先导化合物的潜力。Gan等[44]研发了一种用于治疗乳腺癌的基于天然产物雷公藤红素(CST) 的PROTAC(图4-4),该PROTAC能够通过内源性泛素-蛋白酶体系统选择性地降解肿瘤细胞中的GRP94 和CDK1/4,显著抑制细胞增殖和迁移,并通过凋亡途径诱导细胞凋亡,体内给药实验表明其能安全有效地抑制肿瘤生长。吴茱萸二胺,一种从吴茱萸果实中提取的天然五元杂环生物碱,因其抗菌、抗炎和抗氧化等多种生物活性而闻名。为探索该化合物的分子靶点并增强其抗肿瘤活性,Chen 等[13] 设计合成了吴茱萸碱的PROTAC衍生物(图4-5),研究表明该衍生物在体内外均能有效降解RNA核酸外切酶 4(REXO4),展现出高效的乳腺癌治疗活性和较低的毒副作用。同时,该PROTAC对REXO4 mRNA水平几乎无影响,表明 REXO4 是其直接靶点。He 等 [45] 报道了一系列新的 AXL PROTAC(AXL,一种受体酪氨酸激酶,图4-6),在 MDA-MB-231 TNBC 细胞中能有效消耗 AXL, DC50 值为 5 nmol/L。与相应的激酶抑制剂相比,该化合物对TNBC细胞的AXL信号激活、细胞增殖、迁移和侵袭均有显著改善。Li等[46]基于天然产物灵芝酸 A(GAA)对 MDM2 的亲和力,设计并合成的 V9 PROTAC 和 V10 PROTAC(图4-7)均可与 MDM2 结合并通过泛素-蛋白酶体系统降解MDM2 蛋白,对乳腺癌细胞具有较强的抗增殖作用。其中V10 对MDA-MB-231 细胞(TNBC)的选择性是GAA的 5 倍,有望成为未来TNBC治疗的新型先导化合物。Liu等[47]开创性地报道了叶酸 PROTAC(图4-8)的设计,该分子通过三唑连接臂将叶酸与ARV-771 偶联,凭借FOLR1 介导的内吞途径进入细胞内部,并在水解酶的作用下被激活,从而恢复其诱导蛋白质降解的能力。此外, Chen 等 [48] 研发了首个基于 CRBN 的叶酸 PROTAC,其中二硫键在谷胱甘肽(GSH)的作用下发生还原断裂,释放出针对ALK的活性分子MS4048 PROTAC(图4-9)。汉黄芩素被确认为细胞周期蛋白依赖性激酶 9 (CDK9,IC50 值为 190 nmol/L)的高效抑制剂,揭示了其作为抗癌作用潜在治疗靶点的价值[49-50]。在此基础上,Bian等[51]合成了一系列以汉黄芩素为骨架、针对CDK9 的PROTAC分子。通过人乳腺癌细胞MCF-7 的Western blot分析发现,与广泛抑制CDK家族的汉黄芩素不同,汉黄芩素PROTAC分子(图4-10)能够特异性地降解CDK9,并对过表达CDK9 的癌细胞展现出显著的细胞毒性。四羟黄酮作为一种天然产物及高效的BRD4 选择性抑制剂,在 4T1 和MC38 细胞构建的小鼠肿瘤模型中,展现了良好的肿瘤抑制活性。 Kim等[52]则利用BRD4 的抑制剂 3’,4’,7,8-四羟黄酮设计了一种BRD4 PROTAC,通过持续下调BRD4 水平,有效抑制乳腺癌进展。从印楝叶中提取的萜类天然产物Nimbolide在乳腺癌治疗中展现出了强大的抗癌活性[53]。近期,Gong等[54]通过将Nimbolide与BET 家族抑制剂 JQ1 偶联,成功合成了一系列 Nimbolide 基PROTAC分子。其中,XH2 PROTAC(图4-11)对 BRD4 表现出卓越的降解活性,甚至超过了已知的基于JQ1 的MZ85 PROTAC。
图3针对不同分子分型乳腺癌的PROTAC分子
相比陆地生态系统,海洋生物因总量庞大、物种多样性丰富、资源可再生及成药潜力显著,成为新药研发的关键资源[55-57]。历经约 60 a研究,科研人员已自海洋中鉴别出约 40 000 种潜在药物活性成分(MNPs)。相较于陆生药物,海洋生物新药发现率更高,成药性为陆源生物的 6 倍。目前,逾 1 500 种MNPs进入临床前研究,超 20 种海洋新药进入临床试验,预示其为新药研发的重要方向。在乳腺癌治疗方面,也不乏海洋天然产物的例子。2021 年,Ahmed等[58]发表了 1 篇关于治疗乳腺癌海洋肽的综述,其中Symplostatin 1(图5-1,IC50s=0.15 nmol/L),HTI-286(图5-2) 等海洋来源的肽已被证实在体内外均有治疗乳腺癌的效果。 2023 年 Hussain 等 [59] 概述了上百种潜在的海洋次级代谢产物,这些从海洋来源如海绵、海洋海藻、被囊动物、海洋细菌和真菌中分离出来的次级代谢产物对许多乳腺癌细胞具有细胞毒性。譬如Crambescidin 800 (图5-3),Renieramycin M(图5-4),Aragusterol A(图5-5),Dictyostatin-1(图5-6) 等 IC50s 仅为纳摩尔级别。其中,从红树林相关真菌焦曲霉获得的一种具有蛇胆素骨架的海洋倍半萜 6-epi-ophiobolin G(图5-7),是一种潜在的ERα降解剂,对MCF-7 癌细胞具有抑制作用[60]。基于海洋天然产物软海绵B通过结构简化所筛选出的类似物,研发出了Eribulin mesylate(图5-8),是一种非紫杉烷类微管动力学抑制剂。目前,Eribulin mesylate作为老年HER/2 阴性转移性乳腺癌(MBC) 患者的二线治疗药物 [61]。基于海洋天然产物设计 PROTACs是一种有前景的策略,有助于加快海洋药物在乳腺癌治疗中的研发进程。
图4基于天然产物治疗乳腺癌PROTAC分子
5 小结与展望
本文系统回顾了 PROTAC 技术在乳腺癌治疗领域的应用进展,并针对不同分子分型乳腺癌所对应的关键靶点总结了相应的 PROTAC 分子。研究结果表明,这些PROTAC分子在细胞水平上能有效降解目标蛋白,部分化合物在动物模型中也显示出了积极的治疗效果,为解决乳腺癌治疗中的耐药性问题提供了新的思路。PROTAC技术的快速发展已推动了一系列药物的研发进程。目前,至少有 20 种PROTAC药物正在临床试验中,其中化合物ARV-471 已被FDA授予单药治疗快速通道资格,用于治疗既往接受过内分泌治疗的ER阳性、HER-2 阴性局部晚期或转移性乳腺癌患者。这一标志性事件不仅为ARV-471 后续的临床研究与应用奠定了坚实基础,更为整个PROTAC技术在乳腺癌治疗领域的进一步拓展提供了极具价值的参考范例与成功经验借鉴。
天然产物作为抗癌药物和先导化合物的重要来源,PROTAC技术的运用显著加快了天然产物新药研发的进程[10,62]。一方面,天然产物为PROTAC药物的设计提供了丰富的结构多样性。另一方面,PROTAC 技术通过与蛋白质组学分析等先进技术的结合,成功对多个潜在的癌症治疗靶点进行了精准验证,从而为乳腺癌新药研发工作开辟出崭新的方向[13,63]。本文汇总了基于天然产物的抗乳腺癌 PROTAC 药物分子设计的研究进展,许多陆生天然产物在PROTAC技术的加持下发展成具有前景的候选药物。放眼成药性更高的蓝色药库,PROTAC技术的引入有望在海洋天然产物中发掘出更多具有优异抗乳腺癌活性的分子,进而开发出更安全、更高效的乳腺癌治疗药物。
然而,PROTAC 技术在展现出诸多优势与巨大潜力的同时,也不可避免地暴露出一些局限性与潜在风险。与传统药物通过占位驱动来影响蛋白质功能的模式截然不同,事件驱动的PROTAC只需提供结合活性,触发靶蛋白与E3 酶结合从而引发高效的靶标降解。在成功降解一个靶蛋白分子后,PROTAC能够从三元复合物中解离并迅速进入下一轮循环被再度利用。这种“催化”性能虽然在理论上为高效降解靶蛋白提供了可能,但也潜藏着风险隐患。倘若PROTAC分子缺乏精准的调控机制或者在组织选择性方面表现欠佳,导致其无限制地降解靶蛋白,可能致使正常组织细胞内的靶蛋白也遭受降解破坏从而引发严重的毒副作用。

图5抗乳腺癌的海洋天然产物和海洋药物
综上所述,PROTAC 技术在乳腺癌治疗领域的应用前景广阔,但也面临着一些挑战和局限性。在未来的研究中,研究人员需要继续优化PROTAC分子的设计策略,平衡其高效降解与安全性问题,充分发挥多学科交叉优势,以实现更精准、安全且高效的乳腺癌治疗。同时,研究人员也需要加强海洋天然产物与 PROTAC技术的结合研究,发掘更多具有优异抗乳腺癌活性的分子,开发出更多安全、有效、经济的乳腺癌治疗药物,为乳腺癌患者带来福音。